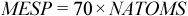| Chapter 10. Electrostatic Potential | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 10. Electrostatic Potential | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Abstract
The ESP program module calculates the expectation values of the electrostatic potential of a molecule on a uniform distribution of points. The resultant ESP surface is then fit to atom centered charges that best reproduce the electron distribution in a least-squares sense. It offers the capability to accurately compute charges in situations where other methods may fail. The keyword ESP is required for all electrostatic potential calculations.
Table of Contents
The set of points defining the default surface is generated according to the algorithm of Connolly.[33] Williams’ method[34] for generating surfaces may be used as an alternative to the default Connolly procedure. Van der Waals radii for the Williams method are included for hydrogen, boron, carbon, nitrogen, oxygen, fluorine, phosphorous, sulfur, chlorine, bromine, and iodine. Van der Waals radii for all elements through chlorine plus zinc are included for the Connolly surfaces. For more information about the surface generation routines in AMPAC™, see the CONNOLLY and WILLIAMS keyword reference pages.
ESP integrals are equivalent to nuclear attraction integrals. The formulae of Obara and
Saika[35] are used in the ESP subroutines. The great majority of the computation time for
a semiempirical ESP calculation is taken in the integral calculation. At the end of the job,
the surface points and electrostatic potential values may be written to file in plain text
format if POTWRT is specified. This
output is written to file jobname.esp
In general, the accuracy of an ESP charge calculation can be enhanced by increasing the number of probe points used for fitting on the Williams or Connolly surfaces. The number of probe points may be adjusted away from default values (determined to be generally adequate by experience) by utilizing the ESP dedicated keywords DEN and NSURF.
|
Enable use of the Connolly surface for the ESP calculation. |
|
|
Specify a different point density for the Connolly surface. |
|
|
Constrain the ESP dipole moment as predicted by AMPAC’s Coulson analysis. |
|
|
Specify the x-component of the dipole moment. |
|
|
Specify the y-component of the dipole moment. |
|
|
Specify the z-component of the dipole moment. |
|
|
Change the number of surfaces used in the Connolly algorithm. |
|
|
Dump out the surface points and electrostatic potential values. |
|
|
Change the base scaling factor in the Connolly treatment. |
|
|
Specify the increment between multipliers for the Connolly surface. |
|
|
Change the scaling factor when using MNDO charges. |
|
|
Specify basis set to “deorthogonalize” the semiempirical density matrix. |
|
|
Specify basis set to “deorthogonalize” the semiempirical density matrix. |
|
|
Average charges which should have the same value by symmetry. |
|
|
Specify surface generation procedure of Donald Williams. |
The implementation of ESP in AMPAC has been generalized to handle UHF and/or CI wavefunctions as well as the more regular RHF solutions. ESP will now also handle charged species. Also, rather than setting the maximum number of probe points available for fitting the surface as a constant (MESP), this value is computed dynamically at runtime time according to the following equation:
where NATOMS is the number of atoms in the calculation.
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 9. Eigenvector Following |  |
Chapter 11. Configuration Interaction |