| Chapter 11. Configuration Interaction | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 11. Configuration Interaction | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Abstract
AMPAC™ offers users a vastly improved and expanded configuration interaction (CI) capability when compared to other semiempirical programs. Given the special keywords and preselection techniques that have been implemented, AMPAC can now perform both extremely complex, yet efficient CI calculations.
Table of Contents
The general-purpose semiempirical models available in AMPAC were originally developed for the efficient prediction of ground state molecular properties at equilibrium geometries. This is where the models were parameterized and where the vast majority of data exists. The level of theory upon which these models are based is the Hartree-Fock SCF method, restricted or unrestricted, which has proved sufficiently accurate to reproduce and predict ground-state properties of most “ordinary” organic molecules and many molecules containing only main-group elements. For excited state geometries and properties, some open-shell systems, atoms or molecules containing metals (e.g., transition metal complexes), properties which involve excited states (e.g., UV/visible spectra) and unusual geometries (e.g., transition states corresponding to weakly avoided crossings) the Hartree-Fock level of theory is often inadequate at best. The need for efficient semiempirical methods capable of handling these cases has long been recognized, especially for larger molecules. Dedicated methods parameterized specifically for some of these cases (e.g., UV/visible spectra) have been proposed and implemented by others. In AMPAC, the approach taken is to use a post-SCF configuration interaction (CI) method, starting with a semi-empirical SCF wavefunction already available. In semiempirical models, some of the “non-specific” electron correlation is absorbed by the parameterization and a crude large-scale CI is usually meaningless. Therefore, an “adapted” CI capable of modeling primarily the “specific” electron correlation necessary to describe excited states and open-shells systems is usually preferable.
An AMPAC N-electron wavefunction Ψ is always initially defined to be a single-determinant SCF wavefunction ΨSCF, i.e., a determinant ψ of orthonormal, variationally determined spin orbitals (SOs) χ. For RHF, the spatial components (MOs) φ of the SOs are restricted to be identical in pairs of alpha and beta electrons (or “half-electrons” of fractional charge for open-shell RHF) during the SCF, whereas this is not the case in UHF. In some RHF cases, it is desirable or necessary (e.g., open-shell RHF, calculation of UV/visible spectra, etc.) to go beyond SCF to a more sophisticated Configuration Interaction (CI) method (CI cannot be used with UHF), where ψ = ψCI is one of many possible variationally determined linear combinations of determinants ψ. The set of determinants ψ combined to form ΨCI is called an N-electron basis (in contrast, to the one-electron basis of Slater orbitals used to expand the SOs) and includes a reference determinant ΨRef (usually ΨSCF) as well as a set of “excited” determinants obtained by moving one or more of the electrons from the occupied SOs of ΨRef to corresponding virtual SOs. For open-shell RHF, the reference wavefunction ΨRef is not ΨSCF since it contains fractionally occupied SOs, but rather a determinant obtained by filling the SOs of ΨSCF in the standard way using the Aufbau principle. In AMPAC, a generic single determinant ψ is referred to as a “microstate”. A general expression for ΨCI can be given by:
where the “C”s are the linear coefficients to be determined, ψia is the determinant resulting from moving an electron from occupied SO χi to virtual SO χa, ψia,jb is the determinant resulting from moving electrons from occupied SOs χi and χj to virtual SOs χa and χb respectively, etc. The sums involving i, j, k, ... are over some subset of occupied SOs in ψRef while the sums involving a,b,c,... are over some subset of virtual SOs in ψRef. Together, these SOs define the CI-active MOs, or the “active space”. A set of CI wavefunctions and corresponding energies can be variationally determined (coefficients C optimized, orbitals fixed) by solving the matrix eigenvalue equation resulting from differentiating the standard Hamiltonian energy expression with respect to the elements of the CI coefficient vector C and setting the result to zero:
where H is a semi-empirical Hamiltonian matrix over microstates (Hpq = <ψp|H|ψq >) and V is the overlap matrix over microstates (Vpq = <ψp|ψq>).
Members of the set of CI wavefunctions satisfying Equation (11.2) are called “CI eigenstates” and are labeled here by their root number in order of increasing energy as Ψ[R], starting with R = 1 for the lowest energy CI eigenstate Ψ[1].
A microstate ψ with Nα alpha electrons and Nβ beta electrons is an eigenfunction of the operator Ŝz (the z-component of the total electron spin angular momentum operator) with eigenvalue Sz:
For an N-electron system, the set {Sz} of possible values for Sz is:
A microstate ψ is not an eigenfunction of the operator Ŝ2 (the square of the total electron spin angular momentum operator) unless it has a closed-shell configuration (all MOs doubly occupied or empty) or a high-spin open-shell configuration (all singly-occupied MOs have parallel spin):
A “spin adapted microstate” η[S,Sz] is a linear combination of microstates that is defined to be an eigenfunction of both Ŝz and Ŝ2:
For example, a microstate with two singly-occupied MOs with opposite spin (Sz = 0) is not an eigenfunction of Ŝ2, but the combination of this microstate with the corresponding spin-flipped microstate is an eigenfunction of Ŝ2, with quantum number S = 0.
For an N-electron system, the set of possible values for S are:
For a given value of S, the set of possible values of Sz are:
The “spin multiplicity” SM corresponding to S is given by:
The spin multiplicity indicates the number of possible values for Sz, and therefore the degeneracy of a spin adapted microstate with total spin quantum number S. For “singlets” (SM = 1, S = 0), there is only one possibly value for Sz: Sz = 0. For “doublets” (SM = 2, S = 1/2), there are two possible values for Sz: Sz = -1/2 and Sz = 1/2. For “triplets” (SM = 3, S = 1), there are three possible values for Sz: Sz = -1, Sz = 0 and Sz = 1.
Since exact eigenstates of the non-relativistic Hamiltonian (modeled by the semi-empirical Hamiltonian H) are pure spin states, i.e., eigenfunctions of both Ŝz and Ŝ2, it useful to constrain the CI eigenstates to be as well. This can be achieved if, instead of using just the “raw” microstates ψ as the N-electron basis prior to solving Equation (11.2), the N-electron basis is defined in terms of spin adapted microstates η.
In terms of a set of spin adapted microstates η[S,Sz] with spin quantum numbers S and Sz, a corresponding pure spin state CI eigenstate Ψ[R,S,Sz]CI is given by:
In AMPAC, the CI eigenstates generated are
always expanded according to Equation (11.13)
and so they are pure spin states. In addition, for efficiency only one
member of a degenerate set of spin adapted microstates is ever used. By
default, this is the one with the smallest non-negative value of
Sz (0 for even-electron systems, 1/2 for
odd-electron systems), but this is modifiable using the keywords
SZ=n or
MICROS=n.
Thus, the CI matrix equations to be solved are:
where H[R,S,Sz]pq = <ηp[S,Sz]|H|ηq[S,Sz]> and Vpq = <ηp[S,Sz]|ηq[S,Sz]>.
Combining equations Equation (11.7) and Equation (11.13), Ψ[R,S,Sz]CI can be expressed directly in terms of the microstates ψm[Sz] with coefficients Dm[R,S,Sz] as:
In the AMPAC output files, it is always the microstate coefficients Dm[R,S,Sz] which are printed.
While there are many CI eigenstates which can be calculated (the number
can be specified using the keyword
CISTATE=n),
AMPAC considers one of them to be the “primary”
CI eigenstate whose energy hypersurface will be followed during geometry
optimizations and which will be used as the reference for all property
calculations. The other n - 1 CI eigenstates requested by
CISTATE=n are considered “secondary” CI
eigenstates, for which some properties are calculated and printed,
typically at one or more optimized geometries of the primary eigenstate,
so their transition properties are non-adiabiatic. By default, the
primary CI eigenstate is the ground state, of any spin
multiplicity, and the secondary eigenstates are all excited states. To
specify a different primary CI eigenstate, use one of the spin
multiplicity keywords
SINGLET,
DOUBLET,
TRIPLET, etc.
and/or
ROOT=n,
where n = 1 refers to the ground state. For example, to use the
second‑lowest energy triplet CI eigenstate (“T2”) as the primary one,
specify TRIPLET and ROOT=2. To use the second‑lowest energy CI
eigenstate of any spin multiplicity as the primary one, specify ROOT=2
without a spin multiplicity keyword.
For the CI eigenstate Ψ[R,S,Sz], the total electron density function ρ[R,S,Sz](r) is expressed in terms of the 2M occupied and virtual SOs χ of ψRef by:
where γmi is the occupancy (0 or 1) of the ith SO for the mth microstate, sz,i = 1/2 for alpha SOs and -1/2 for beta SOs. In terms of the corresponding M MOs, ρ[R,S,Sz](r) is given by:
where γa,mi is the occupancy (0 or 1) of the alpha SO of the ith MO for the mth microstate and PMO[R,S,Sz] is the total one-electron density matrix in the MO basis, with alpha and beta contributions PMO,α[R,S,Sz] and PMO,β[R,S,Sz], respectively. In terms of a basis of L atomic orbitals (AOs) symbolized by ξ, ρ[R,S,Sz](r) is given by:
where PAO[R,S,Sz] is the total one-electron density matrix in the AO basis, with alpha and beta contributions PAO,α[R,S,Sz] and PAO,β[R,S,Sz], respectively.
In AMPAC, when the keyword CIDIP is specified, the dipole moment and Mulliken atomic charges are calculated for both the primary and secondary CI eigenstates from the corresponding density matrices PAO[R,S,Sz]. In general, other one-electron properties which are also available without CI, such as ESP charges, are calculated in CI calculations from PAO[R,S,Sz], but only for the primary CI eigenstate.
The “electron spin density” ρσ[R,S,Sz](r) corresponding to ρ[R,S,Sz](r) is simply the alpha electron density ρα[R,S,Sz](r) minus the beta electron density ρβ[R,S,Sz](r), which, along with the corresponding spin density matrices PMO,α[R,S,Sz] and PAO,β[R,S,Sz] is given by:
In AMPAC, when the ESR keyword is specified, the spin density matrices PMO,α[R,S,Sz] and PAO,β[R,S,Sz] are printed for the primary CI eigenstate along with the net Mulliken atomic electron spins for both primary and secondary CI eigenstates. The net Mulliken electron spin for the Ath atom, σA, is calculated like the corresponding Mulliken atomic electron population except that PAO,σ[R,S,Sz] is used instead of PAO[R,S,Sz]
The transition dipole moment μ[R→n,S,Sz] between CI eigenstates Ψ[R,S,Sz] and Ψ[n,S,Sz] is an important result:
For example, contributions from all available transition dipole moments appear in the “sum-over-states” (SOS) expression for the dynamic polarizability tensor α[R→n,S,Sz](ω), given by Equation (11.26). Individual transition dipole moments are also of interest because they yield information about the UV / visible spectrum of a molecule. The oscillator strength f[R→n,S,Sz]between states ψ[R,S,Sz] and ψ[n,S,Sz] is proportional to the absorptivity of light at a wavelength λ[R→n,S,Sz]:
where K is a constant. By default, AMPAC
writes the transition dipole moments
μ[R→n,S,Sz], transition wavelengths
λ[R→n,S,Sz] and oscillator
strengths f[R→n,S,Sz] between the primary
eigenstate Ψ[R,S,Sz] and all of the
secondary CI eigenstates Ψ[n,S,Sz]. In
AMPAC, the number of CI eigenstates to
calculate, including the primary CI eigenstate, can be specified using
the CISTATE=n
keyword (some of these will have a different total spin quantum number S
than the primary eigenstate and so their corresponding transition dipole
moments vanish).
The “sum-over-states” (SOS) expression for the dynamic polarizability tensor α[R→n,S,Sz](ω) for the CI eigenstate ψ[R,S,Sz] is given by:
where ω is the external electric field frequency (in energy units)
and the sum is over all possible CI eigenstates different from the
primary eigenstate, but having the same S and Sz
quantum numbers. In AMPAC,
α[R→n,S,Sz](ω)
will be calculated and written to the AMPAC
output file when the keywords
DYNPOL or
DYNPOL=n.nnnn
are specified. Note that the keyword
CISTATE=n
has no influence on the calculation of dynamic polarizabilities, and
vice versa, but the number of possible CI eigenstates (determined by the
active space and hence the number of final microstates) does.
The set of occupied and virtual MOs whose corresponding SOs are allowed
to exchange electrons in ΨRef to form new
microstates ψ are called the CI-active MOs or the “active space”.
The choice of active space is one of the most crucial, and sometimes
difficult, steps in a CI calculation, both computationally and in terms
of physical results. Given this importance, the CI-active MOs are
usually specified along with the keywords which invoke CI, possibly
together with the
RECLAS(n,m)
keyword and its associated MO permutation data. For example,
C.I.(5,8) means “do a
CAS-CI using MOs 5,6,7 and 8 as the CI-active MOs”. It is essential
that all or none of the members of a degenerate set of MOs be included
in the active space. By default, AMPAC will
abort if this is not the case. The keywords,
CIGAP=n.nnnn and
CI-OK can be used
to alter the definition of MO degeneracy and to allow the active space
to contain an incomplete set of degenerate MOs. By default, all of the
MO energies are printed to the AMPAC output
file. The keywords VECTORS and
ALLVEC can be
used to print both the MO energies and AO coefficients to the
AMPAC output file for inspection. This
information is also present in an AMPAC
visualization file so that MOs can be visualized with AMPAC’s GUI.
It is important to know the order in which the SCF MOs occur and their
corresponding labeling. For a system with M MOs, the MOs are ordered from
1 to M by increasing occupancy, i.e., first doubly-occupied MOs, then
partially occupied MOs and finally unoccupied (virtual) MOs. This order
usually coincides with increasing MO energy for the entire list from 1 to
M, but within each subset of the same occupancy the order always coincides
with increasing energy.
For RHF open-shell calculations, the SCF calculations in AMPAC
are done using the “Half-Electron” method instead of the ROHF (Restricted Open-Shell Hartree-Fock)
method used by others. In the “Half-Electron” method, the usual
“spin-less” closed-shell RHF SCF formalism is used to calculate
ΨSCF, except that instead of N / 2 doubly
occupied spatial MOs there are assumed to be N / 2 - n (N even) or
N / 2 + 1 - n (N odd) doubly occupied MOs and m MOs with
fractional occupancies which sum to n, where n is the number of open-shell electrons . When the
OPEN(n,m)
keyword is specified, the m open MOs have an equal occupancy of (n/m). When the
SCFCI(n,m1,m2,r) keyword
is specified, the set of m = m1 + m2 open MOs consists of a group of m1 MOs each with an occupancy of (nm1)/(m1+rm2)
and a group of m2 MOs each with an occupancy of (nrm2)/(m1+rm2).
A fractionally occupied MO in the “Half-Electron” method may be
thought of as being occupied by two “half-electrons” of opposite spin
and with a charge equal to half the occupancy of the MO, e.g., (n / 2m) when
OPEN(n,m) is used.
This leads to an energy expression which
is similar to Roothan’s multiconfiguration open-shell SCF energy
expression after spurious coulomb and exchange energies arising from the
interaction between “half-electrons” are subtracted out. In
AMPAC, however, the energy calculated using
the “Half-Electron” method is never used, since it is non-variational,
but the corresponding set of SCF MOs are, either in a “minimal”
CAS-CI calculation involving all of the partially occupied MOs of
ΨSCF as the active space if CI is not
otherwise invoked, or more generally in any specified type of CI
calculation. While the fractionally occupied SOs of
ΨSCF determine the active space of
corresponding MOs, the reference wavefunction
ΨRef used from an open-shell RHF calculation is not
ΨSCF but rather a determinant obtained by
filling the SOs of ΨSCF in the standard way
using the Aufbau principle. It is important to note that, in general,
the number of open-shell electrons to assume for the SCF should be
specified explicitly using one of the keywords
OPEN(n,m),
BIRADICAL,
EXCITED or
SCFCI,
otherwise AMPAC will assume the minimum number
of open-shell electrons (0 for even-electron systems and 1 for
odd-electron systems) for the SCF. The spin-multiplicity keywords
(e.g., SINGLET,
DOUBLET,
TRIPLET, etc.)
are not used in RHF
until the CI portion of the calculation. Thus, for the oxygen molecule,
OPEN(2,2) should be
specified even if TRIPLET
is also specified.
Given ψRef and a corresponding active space,
a definition of which microstates to generate and potentially use for
the expansion of the CI eigenstates is necessary. In the “Complete
Active Space” method (CAS-CI), specified by
C.I.=n or
C.I.(n,m) and
the default when CI is only implied by
OPEN(n,m), all possible microstates which can
be generated by permutations of the electrons among the SOs within the
active space are potentially used. In the “CI Singles” method (S-CI),
specified by
SC.I.=n or
SC.I.(n.m),
all possible singly-excited microstates
ψia are potentially used. In the “CI
Singles and Doubles” method (SD-CI), specified by
SDC.I.=n or
SDC.I.(n,m),
all possible singly-excited microstates
ψia and doubly-excited microstates
ψia,jb are potentially used. In the “CI
Singles, Doubles and Triples” method (SDT-CI), specified by
SDTC.I.=n or
SDTC.I.(n,m),
all possible singly-excited microstates
ψia, doubly-excited microstates
ψia,jb and triply-excited microstates
ψia,jb,kc are potentially used. The
initial set of microstates is referred to here as
{I}MS.
The size of {I}MS grows very rapidly (combinatorially) as the size of the active space increases, especially when CAS-CI is used. (For a CAS-CI involving 10 electrons and 10 CI-active MOs, the number of possible microstates is over 60000, after spin degeneracies are excluded.) In some cases, all of {I}MS should be used, if possible. If this is not the case, whether due to resource limitations and / or to avoid “over-correlating” the already partially correlated, semi-empirically calculated ground state energy, then some means of efficiently selecting the most important “final” set of microstates, referred to here as {F}MS, from {I}MS is necessary. Typically, only a relatively small “target” set of the possible CI eigenstates, {R}ES, are of interest. For example, {R}ES might be composed of the singlet ground CI eigenstate and the first excited singlet and triplet CI eigenstates. {R}ES can usually be characterized in terms of relatively large contributions from a small subset of “germ” microstates {G}MS = {G0, G1, G2, ?}MS, where G 0≡ ψRef roughly corresponds to the ground CI eigenstate R0, G1 to a first excited CI eigenstate R1, etc. While, much of the information relevant to {R}ES is included in {G}MS, the CI eigenstates of {R}ES constructed from {G}MS alone would generally have two significant deficiencies. First, there is generally a lack of specific correlation within the set {G}MS. Second, the excited members {G}MS are lacking in “repolarization” because the SCF orbitals from which {G}MS is generated are obtained from a ground state wavefunction optimization. The objective of the microstate selection procedure used in AMPAC to produce {F}MS is to extract from the enormous list of initial microstates in {I}MS and not in {G}MS, the ones which should contribute most to specific correlation and repolarization. This microstate selection consists of four major steps:
From the initial microstate space {I}MS, keep those J1(≈ 10 × J4, J4 defined below) microstates ψ with the lowest Møller‑Plesset zero‑order energy E0MP[ψ] (sum of occupied SO energies):
where the sums over i and j are over all alpha and beta SOs, respectively, while λ and ε represent SO occupancies and energies, respectively.
From the J1 microstates of step I, choose the J2 (default 100) microstates ψ with the lowest Epstein‑Nesbet (EN) energy EEN[ψ] (semi‑empirical Hamiltonian expectation value).
This set of J2 microstates is the “germ” set {G}MS referred to above.
From {G}MS of step II, determine the J3 (default 30) eigenvectors of the corresponding CI matrix.
From the J1 - J2 “non-germ” microstates ψ which are in {I}MS but not {G}MS, choose the J4 (default 1200) - J2 microstates which make the largest contribution to the following quantity W[ψ]:
At each stage of this microstate selection procedure, the sets of
microstates selected are required to preserve spatial degeneracy, i.e.,
all members of a degenerate set of microstates are kept if there is
space available in the target list, or not kept if there is not space
available in the target list. This is achieved by simple inspection of
the Møller-Plesset zero-order energies, using a degeneracy threshold of
1.0 × 10-4 eV, which is adjustable by
the keyword
CIGAP=n,n.
Of course, this procedure will not cover the case of an active space
containing only a partial set of degenerate MOs. It is important to
remember that either all or none of the members of a degenerate set of
MOs should be included in the active space.
In AMPAC, the above microstate selection procedure can be partially customized by specifying the parameters J2, J3 and J4 using the keywords CIMAX=J4, PERTU=J2 and PERTU(J2,J3).
The following keywords will cause a CI calculation to be done following an RHF calculation:
The following keywords will cause at least a minimum CAS-CI calculation to be done following an RHF calculation. These may also be combined with a keyword which explicitly invokes a larger CI calculation:
The CI-active MOs are always at least least initially defined by the keywords which invoke CI, possibly together with the RECLAS(n,m) keyword. The following keywords modify this definition or have an important bearing on it:
Given an initial set of microstates {I}MS defined by the CI-active MOs, the following keywords help determine the number and nature of the final microstates {F}MS actually used in the CI eigenstate expansions:
The following keywords determine the spin multiplicity SM, CI root and handling of degeneracies (EPS) for the primary CI eigenstate, as well as the number and kind of secondary CI eigenstates calculated:
The following keywords control what kind of extra information is printed to the AMPAC output file or other result files (CIOUT) following any CI calculation:
The following keywords can be used to calculate some extra molecular properties which are specific to a CI calculation:
|
Averaged density matrix in MO basis for the first |
|
|
System has two unpaired electrons. |
|
|
Include n orbitals around the HOMO in the CI manifold. |
|
|
Override degeneracy check. |
|
|
Calculate charges and dipole moments for CI eigenstates. |
|
|
Specify energy gap used to determine microstate degeneracy. |
|
|
Specify the maximum number of microstates. |
|
|
Write details about the CI eigenstates to file. |
|
|
Specify the number of final CI eigenstates to be calculated and printed. |
|
|
Calculate and write transition dipole data between all calculated CI eigenstates instead of just between the primary CI eigenstate and the others. |
|
|
Write details about the CI matrix diagonalization to file. |
|
|
RHF decet state required. |
|
|
RHF doublet state required. |
|
|
Outputs data for dynamic polarizability calculations. |
|
|
Unpaired spin density on atoms will be calculated. |
|
|
First excited singlet state will be optimized. |
|
|
Require use of defined set of prototype MOs. |
|
|
Read final microstates from an ASCII file. |
|
|
All unique two electron integrals over CI-active MOs written to output file. |
|
|
Energies and AO coefficients of CI-active MOs printed to output file. |
|
|
Print information about CI microstates and transitions. |
|
|
Generates only microstates with spin = |
|
|
Maximum charge for generated microstates |
|
|
Constrains the spin multiplicity of the primary CI eigenstate to be n. |
|
|
RHF nonet state required. |
|
|
Expand space of single excitations in a CI calculation. |
|
|
RHF octet state required. |
|
|
Configuration Interaction. |
|
|
Define prototype MOs. |
|
|
Override the default perturbative selection of microstates. |
|
|
RHF quartet state required. |
|
|
RHF quintet state required. |
|
|
Reorder MOs. |
|
|
Propagate initial selection of microstates throughout a geometry optimization. |
|
|
Specify spin state to follow. |
|
|
Defines two sets of open-shell MOs and their fractional occupancies to be used in a “half-electron” RHF SCF calculation preceding a CI calculation. |
|
|
Specify CI-active MOs in a S-CI calculation. |
|
|
Specify CI-active MOs in a SD-CI calculation. |
|
|
Specify CI-active MOs in a SDT-CI calculation. |
|
|
Specify energy gap used to determine eigenstate degeneracy. |
|
|
RHF singlet state required. |
|
|
RHF septet state required. |
|
|
RHF sextet state required. |
|
|
Specify value of Sz. |
|
|
Triplet state required. |
|
|
Indicate that the microstates to be read in are fully consistent. |
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 10. Electrostatic Potential |  |
Chapter 12. Polarizability Methods |
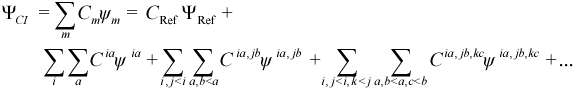
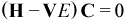
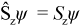
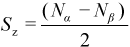
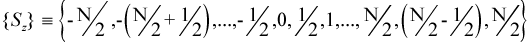
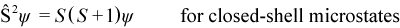
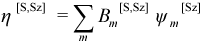
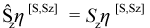
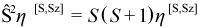
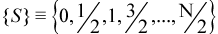
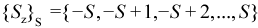
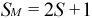
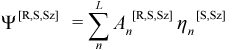
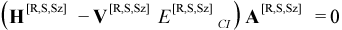
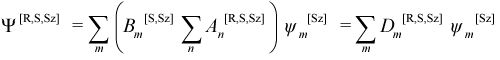
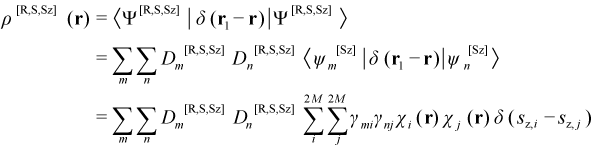
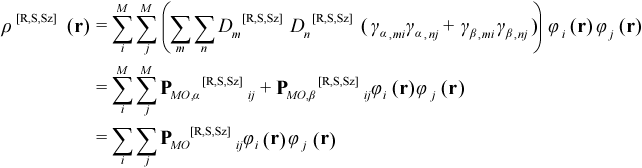
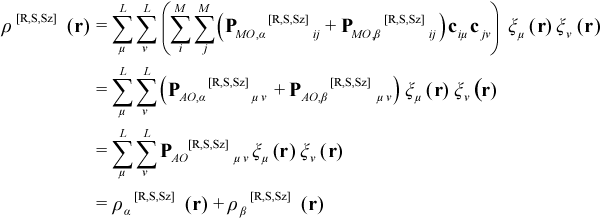
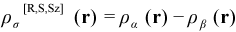
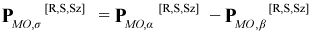
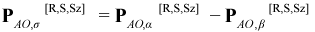
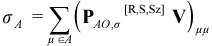
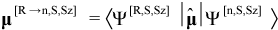
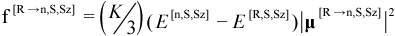
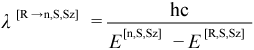
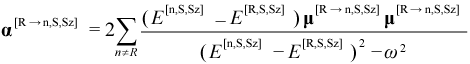
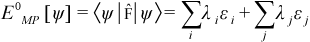
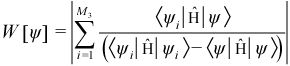
![[Note]](./images/note.png)