| Chapter 34. Use of Partial Charge Sparkles | ||
|---|---|---|

|
Part II. AMPAC™ 10 Examples |  |
| Chapter 34. Use of Partial Charge Sparkles | ||
|---|---|---|
|
|
Part II. AMPAC™ 10 Examples | |
Table of Contents
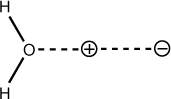
As mentioned in Chapter 5, Presenting Input to the Program, sparkles may be
defined with partial charges to simulate the electrostatic interaction between the molecular
system and some surrounding environment such as a solvent, counterion, or active site. To
illustrate this concept,
gen_sparkles_symmetry.dat
consists of a water molecule polarized by two oppositely charged sparkles in the
following arrangement:
am1 rhf singlet t=1h truste symmetry grad bonds=all WATER WITH TWO PARTIAL CHARGED SPARKLES SPARKLES w/ PARTIAL CHARGES, Na and Cl, T W/ HOURS O 0.000000 0 0.000000 0 0.000000 0 0 0 0 + 1.000000 0 0.000000 0 0.000000 0 1 0 0 0.50H 1.000000 1 128.300000 1 0.000000 0 1 2 0 H 1.000000 1 128.300000 1 180.000000 0 1 2 3 - 2.000000 0 128.300000 0 180.000000 0 1 3 4 -0.50
0 0.000000 0 0.000000 0 0.000000 0 0 0 0 $$ symmetry - constraints
3, 1, 4, 3, 2, 4, 5, $$ end of extra data
|
The “+” sparkle is assigned a charge of 0.50 in the rightmost column. |
|
|
The “-” sparkle is assigned a charge of -0.50 in the rightmost column. |
|
|
This is the extra input section marker for symmetry data. Note, that this marker can be shortened to “$$ symm”. Details of these markers are found in the section called “Extra Input Data”. |
Timestamp: 2011-08-31-12-44-50-0000001038-win64
User Info: John Millam, Nahum,
*******************************************************************************
AM1 CALCULATION RESULTS
*******************************************************************************
* AMPAC Version 10.0.1
* Presented by:
*
* Semichem, Inc.
* www.semichem.com
*
* AM1 - THE AM1 HAMILTONIAN TO BE USED
* RHF - RESTRICTED HARTREE-FOCK CALCULATION
* TRUSTE - MINIMIZE ENERGY USING TRUST REGION METHOD
* SYMMETRY - SYMMETRY CONDITIONS TO BE IMPOSED
* T= - A TIME OF 3600 SECONDS REQUESTED
* BONDS=ALL- PRINT ALL ELEMENTS OF FINAL BOND-ORDER MATRIX
* GRADIENTS- ALL GRADIENTS TO BE PRINTED
* SINGLET - IS THE REQUIRED SPIN MULTIPLICITY
*******************************************************************************
AM1 RHF SINGLET T=1H TRUSTE SYMMETRY GRAD BONDS=ALL
WATER WITH TWO PARTIAL CHARGED SPARKLES
SPARKLES w/ PARTIAL CHARGES, Na and Cl, T W/ HOURS
THE SPARKLE No 2 ACCOUNTS FOR 1 TO THE CHARGE ON THE SYSTEM
BUT HOLDS A CHARGE 0.50000
THE SPARKLE No 5 ACCOUNTS FOR -1 TO THE CHARGE ON THE SYSTEM
BUT HOLDS A CHARGE -0.50000
PARAMETER DEPENDENCE DATA
REFERENCE ATOM FUNCTION NO. DEPENDENT ATOM(S)
3 1 4
3 2 4 5
DESCRIPTIONS OF THE FUNCTIONS USED
1 BOND LENGTH IS SET EQUAL TO THE REFERENCE BOND LENGTH
2 BOND ANGLE IS SET EQUAL TO THE REFERENCE BOND ANGLE
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 O
2 + 1.00000 1
3 H 1.00000 * 128.30000 * 1 2
4 H 1.00000 128.30000 180.00000 1 2 3
5 - 2.00000 128.30000 180.00000 1 3 4
MOLECULAR POINT GROUP SYMMETRY CRITERIA
C2v 0.10000000
SINGLET STATE CALCULATION
RHF CALCULATION, NO. OF DOUBLY OCCUPIED LEVELS = 4
** REFERENCES TO PARAMETERS **
H (AM1): M.J.S. DEWAR ET AL, J. AM. CHEM. SOC. 107 3902-3909 (1985).
O (AM1): M.J.S. DEWAR ET AL, J. AM. CHEM. SOC. 107 3902-3909 (1985).
+ SPARKLE: ALL METHODS.
- SPARKLE: ALL METHODS.
CARTESIAN COORDINATES
ATOM X Y Z
1 O 0.00000000 0.00000000 0.00000000
2 + 1.00000000 0.00000000 0.00000000
3 H -0.61977903 0.78477637 0.00000000
4 H -0.61977903 -0.78477637 0.00000000
5 - 2.00000000 0.00000000 0.00000000
STANDARD DEVIATION ON ENERGY (KCAL) 0.00000055519
STANDARD DEVIATION ON GRADIENT (KCAL/A,RD,RD) 0.00009841 0.00022004 0.00000000
AM1 RHF SINGLET T=1H TRUSTE SYMMETRY GRAD BONDS=ALL
WATER WITH TWO PARTIAL CHARGED SPARKLES
SPARKLES w/ PARTIAL CHARGES, Na and Cl, T W/ HOURS
GEOMETRY OPTIMIZED : ENERGY MINIMIZED
SCF FIELD WAS ACHIEVED
AM1 CALCULATION
VERSION 10.0.1
Aug-31-2011
FINAL HEAT OF FORMATION = 122.807378 kcal
= 513.948877 kJ
ELECTRONIC ENERGY = -505.418009 eV
CORE-CORE REPULSION = 164.749666 eV
TOTAL ENERGY = -340.668343 eV
GRADIENT NORM = 0.004885
RMS GRADIENT NORM = 0.003455
UNSTABLE MODE(S) = 0 ( ESTIMATE )
IONIZATION POTENTIAL = 13.937193 eV
HOMO-LUMO GAP = 17.170605 eV
MOLECULAR WEIGHT = 18.015200
MOLECULAR POINT GROUP = C2v 0.100000
NO. OF FILLED LEVELS = 4 (OCC = 2)
TOTAL NUMBER OF ORBITALS = 6
SCF CALCULATIONS = 8
COMPUTATION TIME = 0.16 SECONDS
FINAL GEOMETRY AND DERIVATIVES
PARAMETER ATOM TYPE VALUE GRADIENT
1 3 H BOND 0.975724 -0.001064 kcal/angstrom
2 3 H ANGLE 129.185666 0.004768 kcal/radian
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 O
2 + 1.00000 1
3 H 0.97572 * 129.18567 * 1 2
4 H 0.97572 129.18567 180.00000 1 2 3
5 - 2.00000 129.18567 180.00000 1 3 4
MOLECULAR POINT GROUP SYMMETRY CRITERIA
C2v 0.10000000
RHF EIGENVALUES
-37.66084 -19.27057 -16.84049 -13.93719 3.23341 4.81211
NET ATOMIC CHARGES AND DIPOLE CONTRIBUTIONS  ATOM CHARGE ATOM ELECTRON DENSITY
1 O -0.4643 6.4643
2 + 0.5000 0.0000
3 H 0.2321 0.7679
4 H 0.2321 0.7679
5 - -0.5000 0.0000
DIPOLE (DEBYE) X Y Z TOTAL
POINT-CHG. -3.776 0.000 0.000 3.776
HYBRID -0.752 0.000 0.000 0.752
SUM -4.528 0.000 0.000 4.528
CARTESIAN COORDINATES
ATOM X Y Z
1 O 0.00000000 0.00000000 0.00000000
2 + 1.00000000 0.00000000 0.00000000
3 H -0.61649701 0.75628624 0.00000000
4 H -0.61649701 -0.75628624 0.00000000
5 - 2.00000000 0.00000000 0.00000000
ATOMIC ORBITAL ELECTRON POPULATIONS
1.85966 1.37307 1.23154 2.00000 0.76786 0.76786
BOND ORDERS AND VALENCIES
1 O 2 + 3 H 4 H 5 -
-----------------------------------------------------------------
1 O 1.892224
2 + 0.000000 0.000000
3 H 0.946112 0.000000 0.946113
4 H 0.946112 0.000000 0.000000 0.946113
5 - 0.000000 0.000000 0.000000 0.000000 0.000000
ELAPSED WALL CLOCK TIME : 0.17 SECONDS
FULL COMPUTATION TIME : 0.16 SECONDS
ATOM CHARGE ATOM ELECTRON DENSITY
1 O -0.4643 6.4643
2 + 0.5000 0.0000
3 H 0.2321 0.7679
4 H 0.2321 0.7679
5 - -0.5000 0.0000
DIPOLE (DEBYE) X Y Z TOTAL
POINT-CHG. -3.776 0.000 0.000 3.776
HYBRID -0.752 0.000 0.000 0.752
SUM -4.528 0.000 0.000 4.528
CARTESIAN COORDINATES
ATOM X Y Z
1 O 0.00000000 0.00000000 0.00000000
2 + 1.00000000 0.00000000 0.00000000
3 H -0.61649701 0.75628624 0.00000000
4 H -0.61649701 -0.75628624 0.00000000
5 - 2.00000000 0.00000000 0.00000000
ATOMIC ORBITAL ELECTRON POPULATIONS
1.85966 1.37307 1.23154 2.00000 0.76786 0.76786
BOND ORDERS AND VALENCIES
1 O 2 + 3 H 4 H 5 -
-----------------------------------------------------------------
1 O 1.892224
2 + 0.000000 0.000000
3 H 0.946112 0.000000 0.946113
4 H 0.946112 0.000000 0.000000 0.946113
5 - 0.000000 0.000000 0.000000 0.000000 0.000000
ELAPSED WALL CLOCK TIME : 0.17 SECONDS
FULL COMPUTATION TIME : 0.16 SECONDS
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 33. Polarizability (KPOLAR, APOLAR) |  |
Part III. AMPAC™ 10 User Reference |