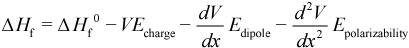| Chapter 12. Polarizability Methods | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 12. Polarizability Methods | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Table of Contents
Polarizability can be defined as the tendency of a molecule’s electrons to be deformed by an applied electric field. The polarizabilties represent the non-linear optical properties of a molecule. This property can be computed by directly adding elements to the Hamiltonian matrix corresponding to the effect of an electric field. The perturbation of the energy operator can be expressed as follows:
This can be used to compute the polarizability tensor (in Å3). In addition to the polarizability, AMPAC computes the first (β) and second hyperpolarizability (γ) values. The β and γ hyperpolarizabilties are respectively the 3rd and 4th order derivatives of the energy (heat of formation) verses the electric field (a Cartesian vector).
KPOLAR is a method for computing a molecule’s polarizabilities contributed by Henry Kurtz. It was designed as a replacement for the now obsolete POLAR method. Using this method, polarizabilties are computed up to gamma hyperpolarizabilties by finite differences at fourth order on both the energy and the permanent dipole. The computed polarizabilities are printed in the inertial frame. (BRUTEKPOLAR is the same as KPOLAR except that results are printed in the genuine Cartesian frame.) While still useful, KPOLAR has largely been superseded by the newer APOLAR.
APOLAR is an alternative to KPOLAR to compute polarizabilities. APOLAR uses finite differences at order three on the variationally-computed permanent dipole, which makes APOLAR cheaper and more precise than KPOLAR. The polarizabilities are computed up to gamma polarizabilties and are printed in both the inertial and genuine Cartesian frames.
This section contains a list of keywords used to invoke polarizability methods.
|
Use Kurtz’s method for computing nonlinear optical properties in the genuine Cartesian frame. |
|
|
Use Kurtz’s method for computing nonlinear optical properties in the inertial frame. |
|
|
Compute nonlinear optical properties using analytic gradient. |
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 11. Configuration Interaction |  |
Chapter 13. Simulated Annealing |