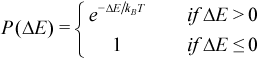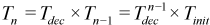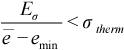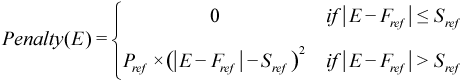| Chapter 13. Simulated Annealing | ||
|---|---|---|

|
Part I. AMPAC™ 10 User Guide |  |
| Chapter 13. Simulated Annealing | ||
|---|---|---|
|
|
Part I. AMPAC™ 10 User Guide | |
Abstract
One of the most difficult problems to solve in computational chemistry is the “multiple-minima” problem, or the location and characterization of geometric minima on a complex multidimensional potential energy surface. Since many chemical phenomena (especially in biological systems) depend on the structural arrangement of the molecule, determining the global minimum energy conformation is an important goal. For complex molecules, there can often be hundreds or thousands of reasonable candidate structures which are almost impossible to generate a priori and adequately explore. The simulated annealing strategy developed here is a heuristic algorithm to address this challenge of finding multiple minima.
Table of Contents
Modern strategies to minimize a function in several unconstrained variables were defined in the 1970s. These methods fall into the general category of “pseudo-Newton” approaches and make extensive use of the gradient of the function, as the only way to retain computational efficiency with large numbers of variables. The minimizers in AMPAC™ (BFGS, EF, DFP, TRUSTE, or NEWTON for energy and TS, TRUSTG, or LTRD for gradient) are all local minimizers in that they converge toward the minimum (or critical point) that is in the most favorable position when related to the starting point. This may not be the global minimum, but only the one that is lowest in energy in a specific region of the potential energy surface. When multiple minima do exist on the potential surface, it is often difficult to locate them directly. A tedious process of trial and error is usually followed by defining a variety of starting points around the area of viability for the optimizable parameters (a grid search). Thus, an open problem in theoretical optimization, the location of the absolute minimum of a function in many unconstrained variables, has direct application to one of the most persistent and annoying problems in computational chemistry, the “multiple-minima” dilemma. In 1983, Kirkpatrick et al. suggested a heuristic answer to this problem, making use of a method of simulation in statistical mechanics coupled with Boltzmann’s law at decreasing temperatures.[36] “Simulated annealing” was the name given the procedure.
A reasonable question for computational chemistry is not necessarily finding the minimum solution of the energy/geometry function defining the molecule, but collecting a variety of possible solutions, i.e. local geometric minima. If properly structured and implemented, the simulated annealing process can be used to collect an entire set of minima. The simulated annealing procedure is very slow in refining the exact location of a minimum, and specialized local methods (much more rapid) are used for exact location. A mixed strategy can be derived in which the approximate locations of the minima are generated by the annealing procedure and the final refinement is carried out from each starting place by one of the local optimization protocols mentioned above.
The theory and terminology behind simulated annealing comes from statistical mechanics (the behavior of systems with many degrees of freedom in thermal equilibrium at a finite temperature). The idea of annealing is to heat an object (e.g. a piece of metal) to a high temperature and then allow the object to slowly cool down. The high temperature allows the atoms to move from their original positions allowing the system to rearrange into a more stable state. As the system cools, it becomes trapped in its new configuration and the system will continue to evolve toward the lowest energy configuration in that local region. Slow cooling may also be followed by a rapid cooling (“quenching”) which removes any residual heat, effectively locking the atoms in their new configuration.
Annealing can used to move a macroscopic system toward its most stable configuration (global minimum) and so has elicited interest in how it can be exploited to solve various types of problems. (In real systems, however, multiple heating/cooling cycles may be required and one is never certain that the true optimal configuration has been achieved.) In typical macroscopic systems (solid or liquid) the density of atoms is on the order of 1023 atoms per cubic centimeter. For such a system in thermal equilibrium, statistical mechanics shows that the behavior of the system is dominated by the most probable behavior and small fluctuations from this average behavior can be neglected. In this type of ensemble, each configuration, defined by a set of atomic positions {ri}, of the system is weighted by its Boltzmann probability factor, exp(-E{ri }/kBT), where E{ri } is the energy of the configuration, kB is Boltzmann’s constant, and T is temperature.
This Boltzmann factor was used to develop the Metropolis step,[37] a simple algorithm that could be used to efficiently simulate a collection of atoms in equilibrium at a given temperature. At each step, a new geometry is generated as a random displacement from the current geometry. The energy at the new geometry is computed and the difference in energy between the current and the new geometries is given as ΔE. The probability that this new geometry is accepted, P(ΔE), is given by Metropolis’ criterion:
Thus, if the new geometry is lower in energy than the current geometry, the step is always accepted. If the step leads to a higher energy, the step will only be accepted if a randomly generated number (between 0 and 1) is less than or equal to the Boltzmann probability factor. At high temperatures, the Boltzmann factor is close to one, so uphill steps (increasing energy) are frequently accepted allowing the system to climb up out of its local valley. At lower temperatures, the Boltzmann factor drops toward zero causing frequent rejection of uphill steps. Thus the system is prevented from escaping its local region and is progressively driven down in energy.
The simple annealing procedure just defined is useful for the global minimum problem but is not well suited for finding multiple minima (or critical points). Also the final stage of annealing converges very slowly and so is inefficient for our purposes. Traditional energy (or gradient) minimizers are much more efficient for the purpose of converging on local extrema. This suggests that the best approach is a heuristic algorithm that effectively mixes the rapid global searching capability of annealing with the efficient local optimization of traditional minimizers. In AMPAC, this mixed annealing strategy is accomplished in three distinct steps.[37]
Each new geometry is generated as a random displacement from the current geometry. Displacements are generated randomly but must be scaled so that they fall within a boundary hypersphere of radius, R. To do this, a random number is generated between 0 and R and the length of the generated displacement is scaled to match this value. By default, a uniform random number is used for this step. Keyword GAUSSIAN forces the use of gaussian random number generator instead. (GAUSSIAN only affects the scaling of the displacement, not the generation of the displacement.) This generated displacement is then added to the existing geometry to produce a new displaced geometry. The energy is computed at this new geometry and is accepted as the new current geometry if it passes the Metropolis criterion (described above). (Note, that the Metropolis criterion can be generalized as will be discussed under the individual simulated annealing methods given later.) When a step is rejected, a new displacement from the current geometry is generated and tested until a successful step is found.
The first step of the simulated annealing phase is to “melt” the system by performing a Metropolis walk on the system at a high initial temperature, Tinit. This walk is continued until the system reaches a steady state (“thermalization”). Subsequently, the temperature is lowered and again allowed to run until thermalization is achieved. Each new temperature, Tn, is determined by multiplying the previous temperature by factor, Tdec, where 0 < Tdec < 1. Thus the nth temperature used is given by:
This process of alternately lowering the temperature and running until thermalization
is achieved is continued until the system “freezes” and no further changes
occur. (Tinit and Tdec are set by keywords
TEMP=n.n
and TLAW=n.n
respectively.)
The boundary radius, R, used in generating displacements is adjusted at each
temperature, Tn. As a rule of thumb, the radius,
Rn, is chosen to give a rejection ratio of approximately 0.5, where
the rejection ratio is the number of points rejected by the Metropolis criterion divided by
the total number of points. This adjustment to Rn can be handled
automatically but is upper bounded by some value, Rmax, to avoid
possible excessive steps. A minimum boundary, Rmin, can also be
specified as fmin * Rmax. If the radius
Rn falls below Rmin, the system is
considered “frozen” and the annealing phase is ended.
(Rmax and fmin are set by STEP=n.n and
STEPCV=n.n respectively.)
The Metropolis walk is continued at a given temperature until “thermalization” is achieved or the maximum number of steps, Nmax, is exceeded. A fairly simple thermalization criterion that can be easily computed is used for this purpose.[38] At every Ncheck points, Eσ (standard deviation of energy), ē (average energy), and emin (minimum energy) is computed for that set of points and tested against our thermalization threshold, σtherm. This thermalization criterion is given as:
(Nmax, Ncheck, and
σtherm are set by NMAX=n, NCHECK=n, and STD=n.n,
respectively.)
Metropolis walks are performed at successively lower temperatures until the system is deemed to be “frozen” (trapped in a local basin) and the simulated annealing phase is ended. The system is deemed frozen if (a) the boundary radius, Rn, is less than Rmin; (b) the temperature falls below a certain threshold (usually Tinit/100); or (c) the final geometry of two or three Markov chains is deemed to be the “same.” By “same” here, we mean that the “distance” between the geometries falls below some threshold.
This Metropolis walk on the PES at successive temperatures generates a series of successive geometries, which are known as Markov chains. These Markov chains can then be used to generate “candidates” for quenching (refinement). Since a typical Metropolis walk visits several minima in the course of its search at a given temperature, and since the walk concentrates on the lower regions of the PES with decreasing temperature, some minima may be visited many times at various temperatures. To avoid excessive minimization during the subsequent quenching phase, all of the candidates are analyzed on the basis of nearly equivalent conformations, accounting for translation, rotation, reflection and for permutations within a set of equivalent nuclei. (This process is also referred to as a “clustering sort”.)
Since geometries close to each other on the Markov chain are generally associated with
the same extrema and quenching is expensive, it is advisable not to quench every geometry.
Instead, all of the Markov chains are divided into segments containing
Ncheck geometries, with only the best conformation in each set
being considered as a candidate for quenching. (Ncheck is set by
NCHECK=n.)
A “distance” between conformations is calculated and candidates are
clustered into closely related groups on this basis. This “distance” measures
the similarity of different conformations taking into account translation, rotation,
reflection, and for permutations within a set of equivalent nuclei.[38] It has been found that only the smaller components of this
“distance” criteria are important, so a band-pass filter is used to eliminate
the larger component contributions. The band-pass filter is defined with central value
BPFref and half-bandwidth of BPFsig *
BPFref. (These values are set by BPFREF=n.n and BPFSIG=n.n
respectively.) Conformations related by a short “distance” are taken as being
equivalent geometries. Candidate geometries from each set are then compared to identify
redundant geometries. If the “distance” between candidate geometries is less
than the value, Filt, the geometries are considered equivalent and one of the geometries is
eliminated as redundant. (Filt is set by FILTER=n.n.)
Each candidate geometry from Stage 2 then undergoes a local minimization (“quenching”) for final refinement of the geometry. This minimization can occur in up to three distinct phases.
By default, TRUSTE (with ANNEAL) or TRUSTG (with TSANNEAL, GANNEAL, or MANNEAL) is used to optimize each geometry to the nearest minimum or critical point. This default can be overridden by explicitly including one of AMPAC’s usual minimizers. For ANNE, an energy minimizer (BFGS, EF, or DFP) can be specified. For TSANNEAL, GANNEAL, or MANNEAL a gradient minimizer (TRUSTG) can be specified. (TS is not recommended since many geometries might be far from a critical point.)
If NEWTON (with ANNEAL) or LTRD (with TSANNEAL, GANNEAL, or MANNEAL) is specified, a second round of quenching is performed but this time using full Hessian methods. Prior to this phase, all of the candidates from phase 1 undergo filtering to remove any equivalent geometries and so reduce the number of geometries to minimize. This optimization step is expensive but can clean up if the initial minimization does poorly.
Typically, a user will freeze less important degrees of freedom throughout the simulated annealing search to focus the search on the more important degrees of freedom. (See Annealing Strategies below.) If keyword WHOLE is specified, all coordinates will be unfrozen and a final full optimization will be performed on each quenched geometry. To have this final optimization done in Cartesian coordinates, the keyword WHOLE=XYZ may be specified. This final phase is referred to as “relaxation.”
To avoid the quenching stage all together, use NOQUENCH. When NOQUENCH is specified, the simulated annealing
job stops just prior to the quenching stage without printing results but producing a
restart file. The job can be finished by adding RESTART in the keyword line and rerunning the job. The final
output of the procedure provides the main characteristics of the minima collected through
the process. Additional output can be obtained by using the PRINT=n keyword.
In AMPAC, simulated annealing has been generalized to include four related strategies, each with their own strengths and weaknesses. The original Metropolis step gives the probability of accepting the step in terms of the change in energy during the step. To apply the Metropolis step and the simulated annealing strategy to more general problems, we can generalize from change in energy to a change in a generalized “cost function.” For chemical systems, this opens the door to using the RMS gradient norm (gnorm) as the cost function in addition to the traditional energy (heat of formation). An additional generalization is that we can add penalty functions to the basic energy or gnorm. TSANNEAL, GANNEAL, and MANNEAL include an energy window penalty term that is directly added to the energy/gnorm used in the Metropolis step. This focuses the annealing search on energies in the window centered at Fref and extends by Sref in either direction. Configurations with an energy outside this window are penalized by a quadratic function with coefficient Pref.
(See annealing keywords, FREF=n.n, SREF=n.n, and PREF=n.n.)
The AMPAC keyword “ ANNE ” represents the standard simulated annealing strategy. The molecule’s energy is used in the Metropolis step and quenching is performed using AMPAC’s energy minimizers. ANNE is useful if one is only interested in minima. (Transition states and other critical points may occasionally still be found but this is uncommon.)
“ TSANN ” is a generalization of ANNE that uses a gradient minimizer instead of an energy minimizer in the final quenching step and thus is useful for locating transitions states rather than just minima. Like ANNE, the molecule’s energy is used in the Metropolis step.
“ GANN ” is a second generalization of ANNE. Like TSANN, it uses a gradient minimizer for the final quenching step. However, it differs from TSANN and ANNE in that the RMS gradient norm (gnorm) is used in place of energy in the Metropolis step. Using gnorm focuses the simulated annealing search on all critical points rather than primarily on just minima.
“
MANN
” is the third generalization of ANNE. For the Metropolis step, MANN uses a
combination of both the energy (like TSANN and ANNE) and gnorm (like GANN). The “cost
function” for the Metropolis step is the minimum of the change in energy and Fctor3
times the change in gnorm. (Fctor3 determines the relative importance of the gnorm compared
to energy in the Metropolis step and is 1.0 by default. This value can be set using the
FCTOR3=n.n keyword.) As with TSANN and GANN, a
gradient minimizer is used in the final quenching step and so is applicable to transition
states and saddle points, not just minima. MANN can be thought of as a merger of both the
TSANN and GANN annealing strategies.
Most annealing algorithms and other multiple-minima search procedures are applied in the context of molecular mechanics (MM). This is because a large number of independent energy calculations usually need to be carried out and MM is the only chemical modeling method efficient enough to do this. To minimize the number of such calculations required using the higher quality, but much slower semiempirical methods, the researcher can apply certain additional constraints to the preliminary search (prior to the use of semiempirical energy calculations), based on his chemical understanding of the problem. These constraints are treated as penalty functions to the genuine search criterion (energy (ANNE, TSANN), gradient norm (GANN), or both (MANN)). Enormous savings and enhanced efficiency can result. For example, in conformational problems involving aliphatic rings, up to 99% of the configurations generated correspond to geometries that are so poor that SCF convergence will be difficult. These conformations are discarded by the annealing procedure at a cost of only 1% of the computer time. The constraints presently implemented are
Explicit boundaries on changing coordinates (Cartesian or internal, with or without symmetry constraints). Such a constraint is actually seen as a periodic condition, as recommended in the original Metropolis algorithm (see annealing keywords LIMIT and AUTOLIMIT).
A severe penalty function is added if an interatomic distance drops below some threshold. This precludes atoms from collapsing onto one another (see annealing keyword PEN1).
A bounded molecular system can be defined in which it is forbidden for any
molecular configuration to have a moment of inertia greater than some defined value (see
annealing keyword PENA=n.n).
Indestructible chemical bonds can be defined by adding a penalty function if a bond
leaves a specific range (see annealing keywords PEN2 and TOL=n).
MANN, GANN, and TSANN have an additional penalty function that defines a specific
energy window that is to be the target of the simulated annealing search. The energy
window is centered at Fref and extends by
Sref in either direction. Configurations with an energy outside
this window are penalized by a quadratic function with coefficient,
Pref. (See annealing keywords, FREF=n.n, SREF=n.n, and
PREF=n.n.)
While the annealing procedure in AMPAC is designed to somewhat automate the search for minima (and other critical points) on a potential energy surface, it is not a “black box.” The judicious use of geometry input and the various penalty functions can make the search both complete in the quantum mechanical potential energy space and efficient computationally. It is neither chemically correct nor necessary to search ALL of the potential energy surface, when only a few geometric variables define all of the meaningful differences between minima and maxima. A few points to note:
Divide the potential energy search into primary and secondary degrees of freedom as the degrees of freedom are expressed in the optimizable geometric parameters. The secondary degrees of freedom can then be stringently constrained using the LIMIT keyword and wide variance can be allowed the primary degrees of freedom.
The TEMP=n.n keyword sets Tinit and can be used to
allow the search to overcome larger energy barriers by increasing the value of
“n.n”. TEMP is expressed in kcal/mol and should be at least twice the
value of the energy barrier to be overcome. The default value is 50 (200 in MANN or
GANN). The annealing process will take substantially longer if TEMP is increased.
The TLAW=n.n keyword allows a wider search of the PES
by slowing the rate of temperature decay, Tdec. A higher value
for “n.n” results in a slower decay rate and correspondingly increases the
computational cost.
The thoroughness with which the PES is searched at each temperature can be
increased by decreasing the value for σtherm (set by STD=n.n). This
may cause the search to locate more remote minima on the PES, at each temperature. Note
that a value of STD that is too small will cause a limited search to require an
inordinate amount of time.
A smaller value of Filt (set by FILTER=n.n) can be used to retain configurations
that are geometrically similar. Too small a value for Filt will result in a large number
of virtually equivalent conformers being passed to the quenching routines. This approach
should be used where conformers are separated by slight differences in geometry.
As the Markov chains are generated, they get sectioned into pieces by the annealing
algorithm. Each piece hopefully refers to a specific minima on the surface. The length
of these sections is governed by Ncheck (annealing keyword NCHECK=n).
A smaller value of Ncheck increases the number of candidates for
quenching, allowing a wider sampling of the Markov chain on the surface but at a higher
computational cost. A large value for Ncheck reduces the
computational cost but focuses the annealing search on a single global minima rather
than multiple minima.
|
Simulated annealing search for geometric minima. |
|
|
Define default preliminary periodic boundaries. |
|
|
Define central value of the band-pass filter. |
|
|
Define half-width of the band-pass filter. |
|
|
Use crude rejection scheme. |
|
|
Determine balance between energy and gnorm (MANNEAL only). |
|
|
Determine equivalency of configurations during the clustering sort. |
|
|
Define central value of the energy range. |
|
|
Simulated annealing search for extrema within an energy range. |
|
|
Use a Gaussian, rather than uniform, random number generator for geometry displacement. |
|
|
Define periodic boundaries. |
|
|
Minimize gradient using full Hessian. |
|
|
Simulated annealing search for minima within an energy range. |
|
|
All points of the Markov chains are written to channel 8. |
|
|
Size of queue to store candidates in simulated annealing calculation. |
|
|
Define interval for producing quenching candidates at each temperature. |
|
|
Minimize energy using full Hessian. |
|
|
Define maximum value of criterion calls at a given temperature. |
|
|
Skip quenching. |
|
|
Define random number seed value. |
|
|
Activate penalty function on the molecule’s moments of inertia. |
|
|
Activate close contact penalty function. |
|
|
Activate conformational penalty function. |
|
|
Activate conformational penalty function within distinct groups. |
|
|
Define the energy window penalty coefficient. |
|
|
Specify half-width of the searched energy range. |
|
|
Define thermalization criterion. |
|
|
Define maximum step size in the annealing search. |
|
|
Define a lower bound for the step size (% of initial step). |
|
|
Starting “temperature” for the annealing procedure. |
|
|
Print extra debugging output. |
|
|
Specify the decay constant in the temperature. |
|
|
Permitted relative variation of a bond length from its initial value. |
|
|
Simulated annealing search for extrema within an energy range. |
|
|
End the quenching steps will full optimizations. |
|
Copyright © 1992-2013 Semichem, Inc. All rights reserved. |
|
|
 |
|
| Chapter 12. Polarizability Methods |  |
Chapter 14. COSMO Solvation Model |